| 深度学习能够有效快速鉴定DDR1激酶抑制剂 | 您所在的位置:网站首页 › ddr1 速度 › 深度学习能够有效快速鉴定DDR1激酶抑制剂 |
深度学习能够有效快速鉴定DDR1激酶抑制剂
|
Deep learning enables rapid identification of potent DDR1 kinase inhibitors 這篇文章作者分別是Insilico medicine創始人亚历克斯·扎沃龙科夫(Alex Zhavoronkov),加拿大多伦多大学化学系的阿兰·阿斯普鲁·古兹克教授及上海藥明康德Li Xing,Tao Guo等科学家團隊负责合成验证化合物的活性。 我们已经开发了用于从头设计小分子的深层生成模型,即生成张量增强学习(GENTRL)。 GENTRL优化了合成可行性,新颖性和生物活性。我们使用GENTRL在21天之内发现了盘状蛋白结构域受体1(DDR1)的强效抑制剂,该靶蛋白与纤维化和其他疾病有关。四种化合物在生化测定中具有活性,其中两种在基于细胞的测定中得到验证。测试了一名主要候选人,并在小鼠中证明了良好的药代动力学。药物发现是资源密集型的,通常需要10-20年的时间,成本从5亿美元到26亿美元1,2不等。 人工智能有望通过促进化合物3,4的快速鉴定来加速这一过程并降低成本。深度生成模型是使用神经网络来生成新数据对象的机器学习技术。这些技术可以生成具有某些属性的对象,例如针对给定目标的活性,从而使其非常适合发现候选药物。但是,关于生成药物设计的例子很少能通过实验验证,涉及合成用于体外和体内研究的新型化合物5-16。 Discoidin域受体1(DDR1)是胶原激活的促炎受体酪氨酸激酶,在上皮细胞中表达并参与纤维化17。然而,尚不清楚DDR1是否直接调节纤维化过程,例如成纤维细胞活化和胶原蛋白沉积,或与减少的巨噬细胞浸润相关的早期炎症事件。自2013年以来,已经发布了至少八种化学型作为选择性DDR1(或DDR1和DDR2)小分子抑制剂(补充表1)。最近,在Alport综合征的Col4a3 – / –小鼠模型中,一系列基于螺二氢吲哚的高度选择性,DDR1抑制剂被证明对肾纤维化具有潜在的治疗功效。因此,更广泛的DDR1抑制剂种类将使人们进一步了解和治疗。我们开发了生成式张量强化学习(GENTRL),这是一种从头设计药物的机器学习方法。 GENTRL优先考虑了化合物的合成可行性,针对特定生物靶标的有效性以及与文献和专利领域中其他分子的区别。在这项工作中,GENTERL用于快速设计对DDR1激酶具有活性的新型化合物。这些化合物中的六个,每个都符合Lipinski的规则1,在46天之内进行了设计,合成和实验测试,证明了该方法在提供快速有效的分子设计中的潜力(图1a)。  Fig. 1 | GENTRL model design, workflow, and nanomolar hits. Fig. 1 | GENTRL model design, workflow, and nanomolar hits.a, The general workflow and timeline for the design of lead candidates using GENTRL. IP, intellectual property. b, Representative examples of generated structures compared to the parent DDR1 kinase inhibitor. c, Generated compounds with the highest inhibition activity against human DDR1 kinase. 为了创建GENTRL,我们将强化学习,变分推理和张量分解组合成一个生成的两步机器学习算法(补充图1)19。首先,我们学习了将化学空间(一组离散的分子图)映射到50个维的连续空间的映射。我们使用张量列格式对学习的流形的结构进行参数化,以使用部分已知的属性。我们基于自动编码器的模型将结构空间压缩到一个分布上,该分布对高维网格中的潜在空间进行了参数化,其节点中的多维高斯呈指数级增长。此参数化将潜在的代码和属性联系在一起,并且可以在使用缺失值没有显式输入的情况下。在第二步中,我们通过加强学习探索了这个空间,以发现新的化合物。 GENTRL使用三个不同的自组织图(SOM)作为奖励功能:趋势SOM,一般激酶SOM和特定激酶SOM。趋势SOM是基于Kohonen的奖励函数,它使用的应用化合物的新颖性进行评分已在专利中公开的结构优先权日期对。 充满新化学实体的神经元会奖励生成模型。一般的激酶SOM是Kohonen图,可将激酶抑制剂与其他种类的分子区分开。特定的激酶SOM从激酶靶向分子的总库中分离DDR1抑制剂。 GENTRL通过依次使用这三个SOM对其生成的结构进行优先级排序。 我们使用了六个数据集来构建模型:(1)来自ZINC数据集的大量分子,(2)已知的DDR1激酶抑制剂,(3)常见的激酶抑制剂(阳性),(4)分子它们作用于非激酶靶点(负组);(5)制药公司已要求保护的生物活性分子的专利数据;(6)DDR1抑制剂的三维(3D)结构(补充表1)。对数据集进行预处理以排除总体异常值,并减少包含相似结构的化合物的数量(请参见方法)。 我们开始在经过过滤的ZINC数据库上训练GENTRL(预训练)(数据集1,如前所述),然后继续进行训练使用DDR1和常见的激酶抑制剂(数据集2和数据集3)。然后,我们启动了强化学习阶段使用前面所述的奖励。我们获得了30,000个结构的初始输出(补充数据集),然后对其进行自动过滤以除去带有结构警报或反应性基团的分子,并且通过聚类和多样性分类减少了所产生的化学空间(补充表2)。然后,我们使用(1)通用激酶SOM和特定激酶SOM,以及(2)基于与DDR1配合的化合物的晶体结构进行药效团建模来评估结构(补充图2和3)。 根据在前面两个步骤(步骤6和7)中计算的分子描述符值和均方根偏差(RMSD),我们使用Sammon映射来评估其余结构的分布。 为了将重点放到较小的分子组中进行分析,我们随机选择了40个结构,这些结构平滑地覆盖了最终的化学空间和RMSD值的分布(补充图4和补充表3)。在40个选定的结构中,有39个可能不在任何已公开专利或申请范围之内(补充表4)。基于综合可及性,选择了其中六个进行实验验证。值得注意的是,我们的方法导致了一些非平凡的潜在生物等位基因替代和拓扑修饰的例子(图1b)。 在选择靶标后的第23天,我们已经鉴定出6个潜在候选对象,到第35天,这些分子已成功合成(图1c)。然后在酶激酶测定法中测试了它们的体外抑制活性(补充图5)。化合物1和2强烈抑制DDR1活性(半最大抑制浓度(IC50)值分别为10和21nM),化合物3和4表现出中等效力(IC50值分别为1μM和278nM),化合物5和6为无效。化合物1和2对DDR1的选择性都比DDR2高(图1c)。此外,与44种不同激酶的那些相比,化合物1表现出相对较高的选择性指数(补充图6)。 接下来,我们研究了化合物1和化合物2的DDR1抑制活性,通过的自磷酸化来测量 U2OS细胞中。这些化合物的IC50值分别为10.3和5.8nM(补充图7)。这两种分子均抑制了MRC-5肺成纤维细胞中纤维化标记物α-肌动蛋白和CCN2的诱导(补充图8)。这些分子还抑制LX-2肝星状细胞中胶原蛋白的表达(纤维化的标志),化合物1在13nM时显示出强效活性(补充图9)。 然后,我们进行了体外微粒体稳定性研究,以表征化合物1和2在人,的代谢稳定性大鼠,小鼠和狗肝微粒体中。化合物1和2的半衰期和清除率值与的半衰期和清除率值相似或更高常规对照分子(补充表5)。还发现化合物2在缓冲液条件下非常稳定(补充表6)。两种化合物均未强烈抑制细胞色素P450,并且两种化合物均显示出良好的理化性质,包括满足Lipinski规则(补充表7和8)。 最后,我们在啮齿动物模型中测试了化合物1。将化合物1静脉内(iv)(10mg kg-1)和口服递送给小鼠(po,15mg kg-1)。两次给药的半衰期相近,约为3.5h(图2a以及补充表9和10)。静脉内给药在初次给药时使血浆峰值浓度为2357ng ml-1,而口服给药导致最高血浆浓度为266ng ml-1 较低,在给药后1 h达到峰值。  Fig. 2 | Pharmacokinetic characterization and structural basis of hit activity. Fig. 2 | Pharmacokinetic characterization and structural basis of hit activity.a, Plasma concentrations of compound 1 in mouse pharmacokinetic study at doses of 10 and 15 mg kg–1 for i.v. and p.o. treatment, respectively. Measure of center is mean; error bars are s.d.; n= 3 biologically independent animals used for each route of administration. b, The rigid alignment of a conformation that best fit the pharmacophore hypothesis and a conformation predicted by quantum mechanical calculations. Superpositions are presented for compound 1, compound 3, and compound 5. Orange, quantum mechanical calculation; yellow, pharmacophore modeling. c, The putative binding mode of compound 1 (IC50= 10 nM) in DDR1 kinase (PDB code: 3ZOS) derived from docking simulations. The receptor is shown in gray; compound 1 is shown as sticks and balls, and key receptor residues that are involved in ligand binding are shown as sticks. Hydrogen bonds are shown as yellow dashed lines. 量子力学分析被用来探索化合物1活性的机理基础。根据药效团模型,化合物1的预测构象与量子力学计算预测的优选和稳定构象非常相似(图2b)。我们提出了化合物1和DDR1之间的“锁和键”熵驱动的结合机制,并通过分子对接进一步表征了这种结合。推定的结合模式表明II型抑制机制(图2c)。总之,化合物1形成多个氢键,并且与DDR1激酶的活性位点残基具有有利的电荷和疏水相互作用。化合物1与ATP位点的互补性可能有助于解释其对DDR1的抑制活性。尽管具有合理的微粒体稳定性和药代动力学特性,但在选择性,特异性和其他药物化学特性方面,此处确定的化合物可能仍需要进一步优化。在这项工作中,我们在不到2个月的时间内设计,合成和实验验证了针对DDR1激酶的分子,而其成本仅为传统药物发现方法1的一小部分。这说明了我们的深度生成模型在成功,快速设计合成上可行,对目标感兴趣的化合物以及在现有知识产权方面具有潜在创新性的化合物的成功应用。我们预计,该技术将作为识别候选药物的有用工具而得到进一步改进。 评论: 最近出版 “ 深度学习能够有效的DDR1激酶抑制剂的快速鉴定 ”周围亚历克斯·黑文罗恩科弗的团队在硅片医药,连同药明康德和多伦多大学,先后获得颇有些关注最近-所以有什么呢? 人工智能“发现了第一个药物”真的发生了吗?让我们更详细地看一下这项工作。 作者使用了“生成张量强化学习(GENTRL)”的实现,其目标功能包括有关目标活动,合成可行性和新颖性的信息。合成了针对激酶DDR1设计的六种化合物,并在生化分析中进行了测试,产生了四种活性化合物(低于10uM,其中一种化合物对DDR1的IC50降至10nM),两种化合物在细胞分析中具有活性。此外,还确定了化合物在小鼠中的药代动力学。 无论如何,让六种化合物中有四种甚至两种,对预期目标都具有“活性”,这肯定不是一个坏的“命中率”。但是新颖性如何?我们不开发抗蛋白质药物,而是打算治疗人 -因此,该化合物的药代动力学,功效和安全性如何? 为了评估这些化合物的新颖性,我刚刚对它们活性最高的化合物(化合物1)进行了75%相似度的ChEMBL搜索,以评估公共领域(或至少在此数据库中)的新颖性:  我们可以看到,该算法重新排列了已知激酶抑制剂的杂环系统,以提出新颖/重新排列的结构。在这种情况下,针对ABL的活性可能外推至DDR1,这是两种具有已知交叉反应性的激酶。(但是,由于我无法自己详细地复制工作流程,因此我也不能绝对确定该结构的起源。)有趣的是,合成(和其他)过滤器还对与已知化合物相似的子结构进行了优先排序-也许合成化学具有相当强的偏见和偏好(当然,这并不是一个全新的发现!)。 除了靶向活性外,作者还评估了该化合物的药代动力学,他们认为这是有利的-但是,这似乎与设计假设无关(即,此处未明确考虑PK)。 我对该文章的欣赏是,它没有声称它以任何方式“发现了药物”, 而不是将某些推文描述为“从事药物发现的AI”。但是,药物需要在体内显示其作用,而这项工作尚未执行(至今!)。不过,这将是合乎逻辑的下一步-甚至更重要的是,这是至关重要的一步,因为临床开发中的大多数药物都由于缺乏功效而失败,而这很难从早期数据(化合物的分布,新陈代谢,靶标参与等,不仅仅局限于与分离的靶标结合。 因此,鉴于体内研究仅包含PK且不包含功效(或广泛的毒物)成分,我可能不会同意AI在Twitter上某些读者的情况下已经执行过“药物发现”的评论 -但它具有我当然同意在短时间内发现新的生物活性化学物质。 可能需要指出的一件事是,本文使用了很多其他早期项目中实际上没有的信息,例如晶体结构数据和有关现有活性化合物的信息。如果可用的信息少得多,该方法将如何在其他目标上执行?看到一个“简单的”基线方法进行比较会很有趣–那么以沼泽标准为基础的虚拟筛选,对接和蛋白质化学计量学建模会如何呢?鉴于所有这些配体和结构信息均可用,因此可以比较容易地将其用于比较-而且,我们真正需要使用一种方法的信息越少,则在实践中将越广泛地适用。 我期待着这项工作的下一步,尤其是进入生物学领域-在预期疗效和安全性方面,以计算机方法解决发现药物的生物学步骤可能会带来巨大的优势,从而减少药物的消耗诊所。 /安德烈亚斯 作者: andreasbenderAndreas Bender /开发和应用化学和生物学数据的新数据挖掘方法,用于药物发现,化学生物学和计算机毒理学/剑桥大学分子科学信息学中心的读者/ Healx Ltd.的联合创始人,用于以数据为基础的药物重用以治疗罕见疾病的方法http://www.healx.io/ PharmEnable Ltd.的联合创始人,用于获取针对困难目标的新化学方法http://www.pharmenable.com 查看andreasbender的所有帖子 最后强烈推荐肖高铿老师的教程,用更简单的软件实现类似的结果。 用SPARK基团替换策略重现深度学习DDR1抑制剂的发现 Cresset 2019-09-05 940 0 4 摘要:本文以DDR1抑制剂Ponatinib为起始化合物,用SPARK对其进行了基团替换计算实验。结果表明,SPARK在不到10分钟的时间内完成了计算,不仅获得了Alex等人采用深度学习生成化合物1的甲基哌嗪衍生物,而且该类化合物被SPARK打分后排序靠前(排名第一)。这说明SPARK比起深度学习在使用上可以更加简单、直接地获得目标化合物。肖高铿/2019-09-05深度学习生成DDR1抑制剂英科智能(Insilico Medicine)Zhavoronkov Alex等人最近在Nature Biotechnology杂志上发表关于深度学习加速DDR1激酶抑制剂发现的文章1。药明康德微信公众号文章《从靶点到候选分子仅需3周!》报道并总结了该文的亮点, 并评论说:“值得一提的是,在人工智能技术与研发人员的协同下,在选定靶点的46天后,新筛选出的分子就完成了初步的生物学验证”。  Figure 1. GeNTRL模型的设计、工作流与nM苗头化合物。a,GeNTRL分子设计总的工作流与时间线. b, 已知的DDR1激酶抑制剂与生成的代表性结构;c, 生成的对人类DDR1激酶抑制活性最强的化合物Figure 1总结了抑制剂发现的流程与时间线;展示了生成的代表性化合物与实验验证的活性最强DDR1抑制剂。总的来说,深度学习确切无疑地加速了药物的发现过程,经过实验测试与验证,化合物1(Figure 2)被认定为最有发展潜力的候选化合物(原文为Lead candidate,我把它理解为candidate)。  Figure 2. Alex等人1用深度学习设计的化合物1 Figure 2. Alex等人1用深度学习设计的化合物1鉴于Alex的化合物1与已知的DDR1抑制剂Ponatinib(Figure 3)在结构上有很深的“渊源”, 有经验的药物化学家认为:化合物1的1,2-苯并异噁唑片段(Figure 2的黄色高亮部分)是Ponatinib苯甲酰基(Figure 3的高亮部分)显而易见的生物等排体,所以化合物1事实上是Ponatinib的me-too开发。视频1演示了基于结构的Me-too计算实验:通过观察Ponatinib与DDR1的相互作用,可以发现C=O作为氢键受体与蛋白ASP784发生氢键作用,将该C=O氧用SP2的N氮代替并环合,可以保留该相互作用同时保留分子的形状不变。  Figure 3. 已知的DDR1抑制剂Ponatinib,黄色高亮部分被生物等排体替换可获得化合物1的衍生物 Figure 3. 已知的DDR1抑制剂Ponatinib,黄色高亮部分被生物等排体替换可获得化合物1的衍生物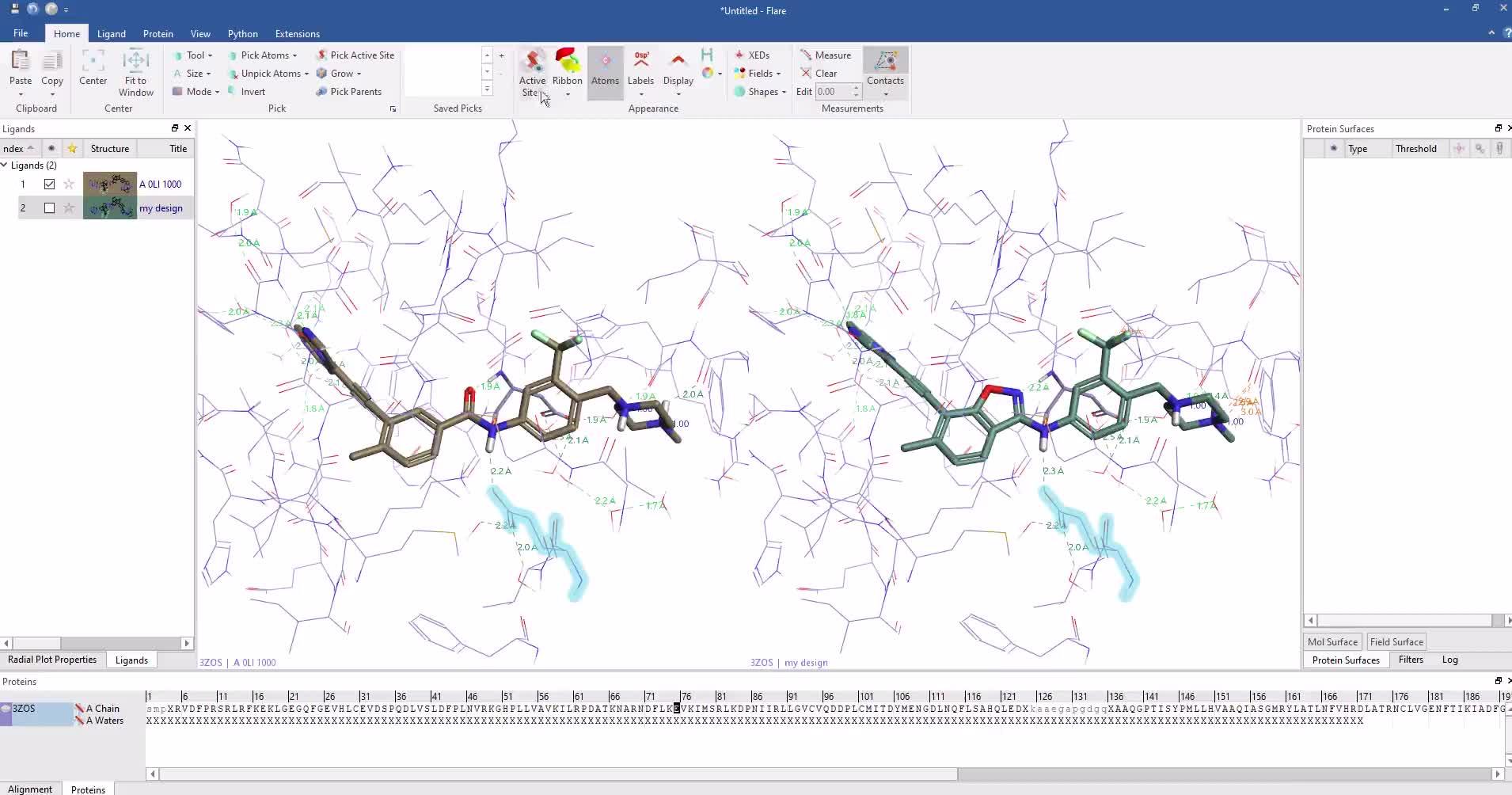 https://www.zhihu.com/video/1160523431912349696 https://www.zhihu.com/video/1160523431912349696视频1:利用化学经验对Ponatinib进行基于结构的me-too计算实验: 通过观察Ponatinib与DDR1的相互作用,可以发现C=O作为氢键受体与蛋白ASP784发生氢键作用,将该C=O氧用SP2的N氮代替并环合,可以保留该相互作用同时保留分子的形状不变。 我感兴趣的是:(1)用传统药物设计技术,比如基团替换策略能否重现Alex等人1的结果?(2)需要多长时间?(3)设计出来的化合物能否排序靠前以便药物化学家们马上关注到该化合物?为此,用SPARK的生物等排体替换策略尝试对DDR1抑制剂Ponatinib进行基团替换,以Alex等人的化合物1为目标考察计算结果,并与大家分享结果。用SPARK对DDR1抑制剂Ponatinib进行生物等排体替换从PDB下载Ponatinib与DDR1激酶的复合物晶体结构(3ZOS),并将Ponatinib作为SPARK基团替换的起始化合物(Starter),基团替换时仅搜索ChEMBL的常见片段库对Ponatinib的苯甲酰基片段(Figure 3黄色高亮部分)进行替换,具体过程见下面视频。 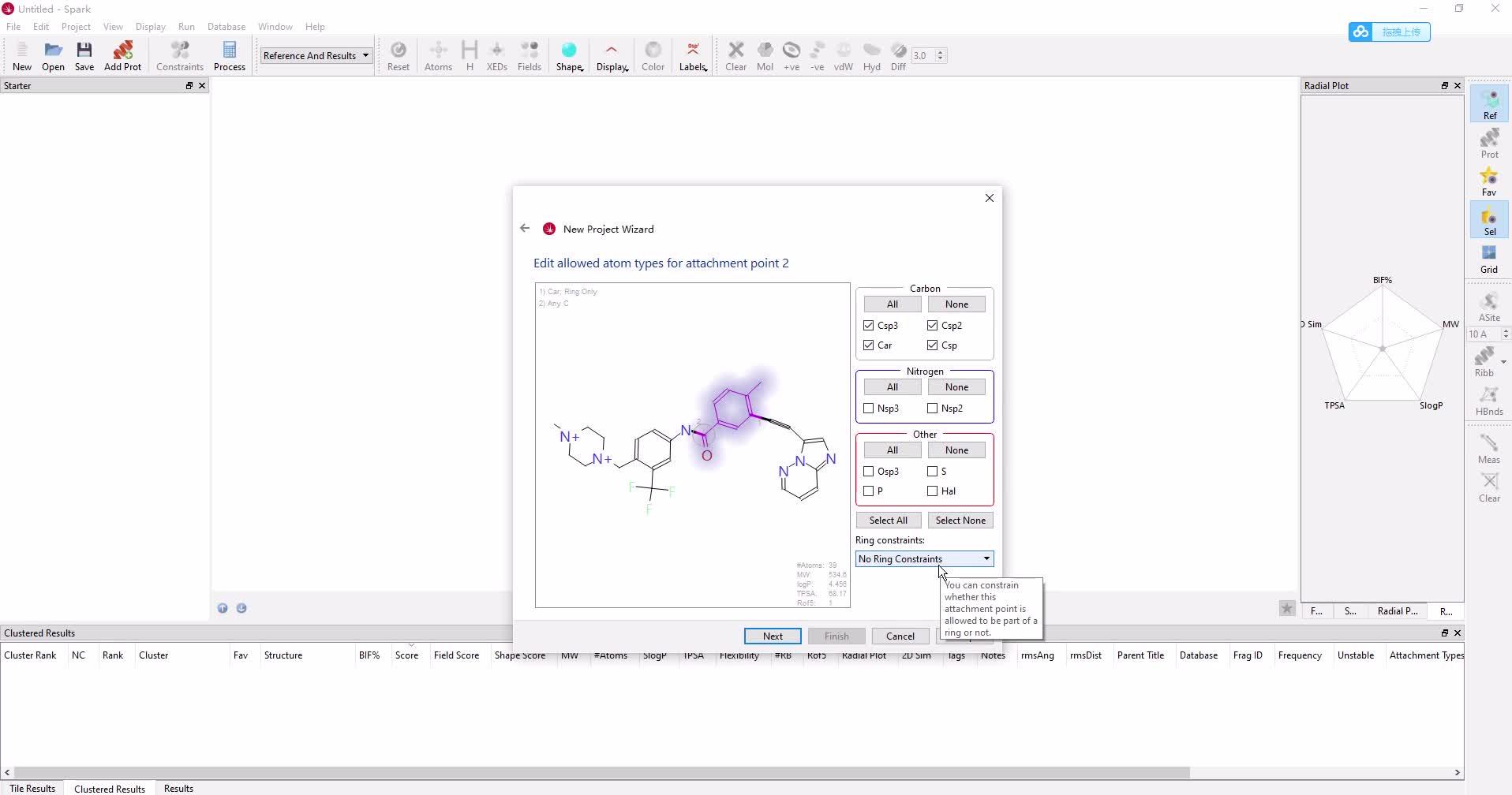 SPARK生成了化合物1的甲基哌嗪https://www.zhihu.com/video/1160523679510536192 SPARK生成了化合物1的甲基哌嗪https://www.zhihu.com/video/1160523679510536192视频2:用SPARK对Ponatinib进行基团替换的实验: SPARK生成了化合物1的甲基哌嗪衍生物以及其它的备选化合物 如视频2所示,我们仅用ChEMBL(Version 23)的115610个常见片段进行了筛选(见Figure 4),计算采用默认值,最大输出500个打分最佳的化合物。整个过程10分钟不到就完成计算、获得计算结果。  Figure 4. 基团替换仅对ChEMBL(Version 23)的115610个片段进行筛选Figure 5所示展示了打分最高的前48个化合物(仅展示替换片段),您会发现Alex等人1化合物1骨架排序第1。  Figure 5. 打分最高的前48个化合物(点击图片切换至大图片)对500个结构进行聚类,共得到214中不同的结构类型,按打分从高到低排序,其中排序第1的类别含有三个化合物正是Alex等人深度学习生成的化合物1,如Figure 6所示。Figure 6也给出了这三个化合物的骨架来源,分别来自CHEMBL523516,CHEMBL3338562与CHEMBL493496。 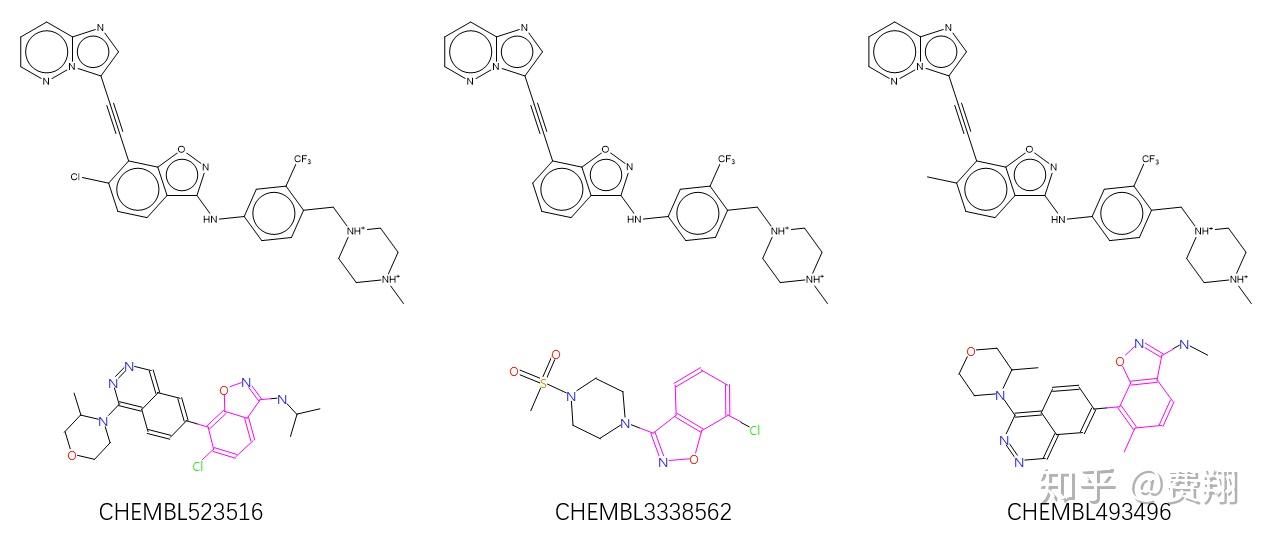 Figure 6. 打分最高的Cluster 1包含三个化合物(上)及其CHEMBL骨架来源(下),三个化合物在全部500个化合物排序分别为1、3与6(从左到右)Figure 6右边化合物是Alex等人的化合物1的甲基哌嗪衍生物,SPARK的基团替换策略重现了Alex等人的深度学习从头设计结果;除此之外,还生成了羰基对位去甲基或氯取代的衍生物,该类化合物被SPARK的形状与场点相似性综合打分排序第一,药物化学家可以马上关注到该类化合物;虽然也需要对SPARK的结果进行谨慎地选择与后处理,但是在这个练习中不需要进行层层地性质过滤、药效团过滤等等操作就富集到了目标化合物。 视频3:Ponatinib及基团替换后化合物与DDR1的相互作用模式 结论本文以DDR1抑制剂Ponatinib为起始化合物,用SPARK对其进行了基团替换计算实验。结果表明,SPARK在不到10分钟的时间内完成了计算,不仅获得了Alex等人采用深度学习生成化合物1的甲基哌嗪衍生物,而且该类化合物被SPARK打分后排序靠前(排名第1)。整个过程没有对化合物进行各种过滤,这说明SPARK比起深度学习结构生成在使用上可以更加简单、直接地获得目标化合物。 关于insilico公司还可以参考这篇文章:看AI如何在技术上,做到药物研发上的创新? - 彩云CSR-肖云的文章 - 知乎 |
【本文地址】